药代动力学
研究内容
研究药物的体内过程和体内药物浓度随时间变化规律。术语
- 机体对药物的处置(disposition)
- 吸收、分布、代谢、排泄(ADME)
- 转运
- 吸收、分布、排泄(ADE)
- 消除
- 代谢、排泄(ME)
- AUC
- 浓度-时间曲线下面积,与吸收量成正比
药物的转运
重点:脂溶扩散、离子障、pH对转运的影响。 转运最基本的是跨膜转运(包括膜动转运即胞吞胞吐)。 绝大多数药物是通过简单扩散,尤其是脂溶性的扩散进行转运的。 给药部位浓度最高,再扩散至作用部位。 影响脂溶扩散的主要因素是脂/水分配系数、浓度梯度、解离度,此外还有膜面积等。 脂溶扩散存在离子障:非离子型药物可以自由穿透生物膜,离子型则被限制在膜一侧。 这是由离子型极性高,水溶性高导致的。 pH值常可改变离子型和非离子型的比例,从而影响其转运。 体内pH值改变不大,故解离度影响药物转运。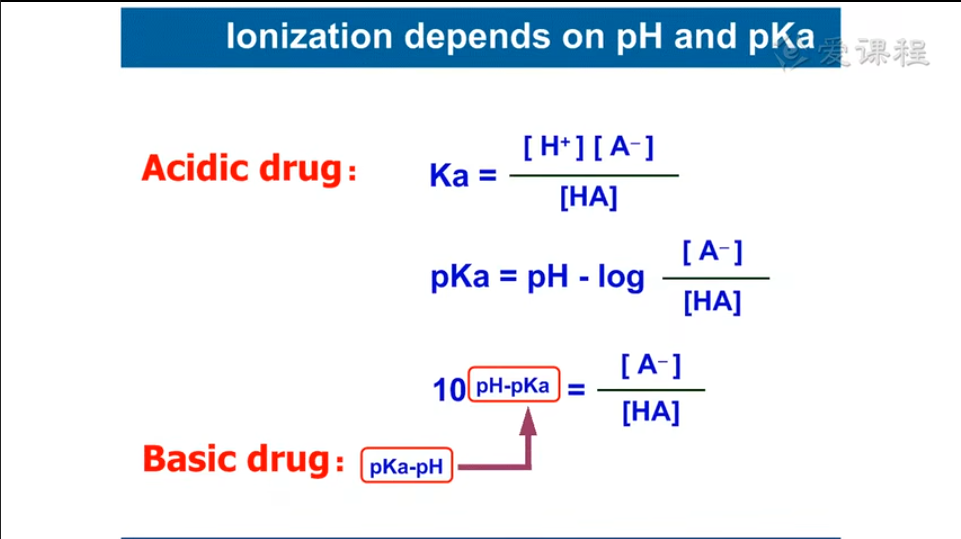
例如,弱酸性药物中毒时,应碱化尿液,抑制其重吸收。 常识:胃液、乳汁、尿液正常情况下均偏酸。
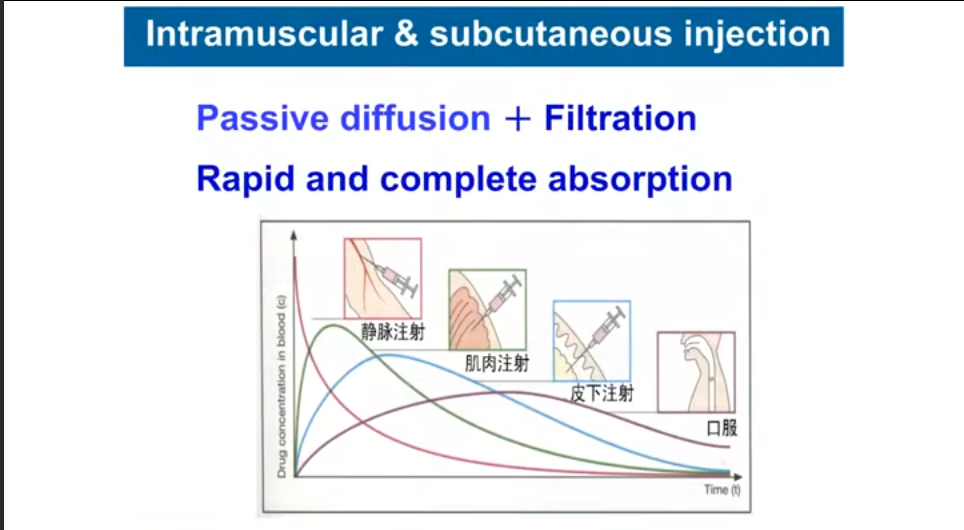
注射给药时,药物直接在细胞间隙扩散,此时主要为水溶性扩散,油剂反而扩散效率慢。
各个过程
吸收
首关效应:- 口服用药
- 药物在肠肝被代谢
- 最终进入体循环的药量减少
首关效应很强的典型药物:硝酸甘油。
肺部吸收面积很大,效率可与静脉注射媲美,且无首关消除。
生物利用度:药物经血管外途径给药后吸收进入全身血液循环的相对量,以AUC表示。 分为绝对生物利用度(静脉给药为分母)和相对生物利用度(标准制剂为分母)。分布
通常在血流丰富的组织和器官,药物的分布速度快而且转运量较多(如脑、肝、肾、肺),随后还可以再分布(redistribution)。
大多数药物可与血浆蛋白形成结合性药物。
性质:- 可逆性=>平衡&血浆蛋白结合率、游离浓度
- 可饱和性
- 竞争性
- 限制其扩散至血管外作用部位
- 延长作用时间
- 相互作用
血脑屏障使得只有脂溶性高的药物才能入脑,极性高或分子量大的药物难以进入脑内。
胎盘屏障对药物无屏障作用。
代谢
生化相关:时相——I相反应(氧化还原水解)、II相反应(结合)
肝中代谢药物最重要的一组酶:细胞色素P450单加氧酶系。CYP3A代谢了50%的药物。
- 酶的抑制
- 抑制剂(如西米替丁)使得酶活性降低(蛋白质层面),延长药理作用(前药相反)。
- 停药敏化,停止一种药之后另一种药未作调整而出现中毒。
- 酶的诱导
- 诱导剂(如苯巴比妥、利福平)使得酶表达增高(基因表达层面),大部分药物代谢加快,首过效应加强,生物利用度降低。
- 前药正相反,需要酶使得其转化为活性形式。
排泄
途径:肾、消化道、呼吸道(尤其是酒精)、泪腺、汗腺。
肾小管可以通过非特异性转运分泌有机阴离子和有机阳离子,经过同一机制分泌的药物可发生竞争性抑制。
血和尿的pH值和药物的pKa可影响药物在肾小管的重吸收。
肝脏、小肠、胆汁之间的循环称为肠肝循环,可延长药物的血浆半衰期和作用维持时间。
房室模型
一室模型:药物在全身各组织部位的转运速率相同或相似,此时把整个机体视为一个房室。
二室模型:从速率论的观点将机体划分为药物分布均匀程度不同的两个独立系统。其中血流丰富以及药物分布能瞬时达到与血液平衡的部分称为中央室;血液供应较少药物分布达到与血液平衡时间较长的部分称为周边室。
其他重要参数
表观分布容积:血浆和组织内药物分布达到平衡时,体内的药物按血浆药物浓度在体内分布所需体液体积。可以据此推测药物在体内的分布情况。
消除半衰期:按一级动力学消除的药物半衰期为一常量(0.693/k);零级动力学消除的药物半衰期则与初始浓度有关(这是显而易见的)。